
| Download File Name | Available at | Size | MD5 |
|---|---|---|---|
| 16.marker.82.xlsx | CarrotOmics | 90.93KB | fc724b1523752b9646c20377ddf10c19 |
Linkage map markers and information.
| Genetic Map | 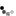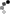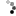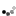 |
|---|
| Name | Description | Units |
|---|---|---|
This map was constructed using an F₂ population L8708×Z020 that segregates for plant height, shoot biomass, and storage root shape. Linkage maps were constructed using the JoinMap 4.1 software (Van Ooijen, 2011). Markers and genotypes which deviated from expected segregation ratios based on a Chi-square test (P < 0.001) were excluded. All linkage groups were obtained at a LOD threshold greater than 10. The regression mapping algorithm was used with Kosambi’s mapping function to calculate the distance between markers (Kosambi, 1943). Linkage groups were achieved by aligning GBS sequences to the carrot genome (Iorizzo et al., 2016) and corresponded to nine chromosomes. After initial mapping, markers defined as having insufficient linkage were flipped to the opposite phase and remapped. Two rounds of the regression mapping algorithm were used to increase the number of loci incorporated into the map. QTL analysis was conducted in R 3.3.2 (R Core Team, 2016) using the R/qtl package (Broman and Sen, 2009). Individuals included 316 F₂ plants from the CA2016 environment. Genotype probabilities were calculated using a step value of one for the entire linkage map and an assumed genotyping error rate of 0.001. Missing genotype data was replaced with the most probable values using the Viterbi algorithm (method = ‘argmax’) in the ‘fill.geno’ function. Multiple QTL mapping (MQM) (Jansen and Stam, 1994) was performed in R/qtl using the ‘mqmscan’ function with an additive model and cofactor significance set to 0.001 (Arends et al., 2010). Cofactors were set at a fixed marker interval of 5 cM. Following scripts developed by Moore et al. (2013), genome-wide LOD significance thresholds were determined for each phenotype using parallel computing on the Open Science Grid (OSG) (Pordes et al., 2007; Sfiligoi et al., 2009). Significance thresholds were based on 10,000 random permutations (Churchill and Doerge, 1994) with the assumed genotyping error rate set to 0.001 and α = 0.01. For each QTL, confidence intervals were determined using the 1.5 LOD drop off flanking the most significant peak of the QTL. | cM |
| Name | Attribution 4.0 International (CC BY 4.0) |
|---|---|
| License Summary | You are free to:
The licensor cannot revoke these freedoms as long as you follow the following license terms:
Notices:
|
| Full Legal Text | https://creativecommons.org/licenses/by/4.0/legalcode |